
How to Set Suitability Criteria for Relative Potency Bioassays
We previously discussed the purpose of system suitability tests and sample suitability tests and gave some examples of each. However, we did not explain how to actually set the limits for these tests: suitability criteria. In this blog we will discuss this and go through a couple of examples of how to set suitability criteria in practice.
There is fairly detailed guidance on how to set limits for suitability tests in Chapter 1032 of the United States Pharmacopeia (USP). Four methods are discussed – we’ll show examples of each of these below. The discussion in the USP is only about parallelism, and we will also use parallelism in our examples below, but in fact these methods can be applied to any suitability test, including both system and sample suitability.
The first two methods (“a” and “b” in the USP) use only “good” historical data, where the assay and samples were behaving as expected (of course, you never really know this for certain, but you have to start somewhere…). For parallelism, a potential source of “good” data are assays where the reference standard was tested against itself, as these should certainly be parallel.
Key Takeaways
- The USP describes four approaches (methods a–d) for setting bioassay suitability limits; methods A and B use only “good” historical runs, method C adds known “bad” runs, and method D focuses on the real-world impact of assay bias.
- Methods A and B are easy to implement and ensure most good assays pass, but may not reliably reject bad assays; method C improves discrimination by explicitly including failed or degraded samples in the limit-setting process.
- Method D sets limits based on acceptable impact on decisions (for example, batch release), but requires a deep understanding of assay behaviour and is therefore rarely used in practice.
- Suitability criteria are not fixed forever; as more data (including problem assays) accumulate, limits and even the chosen method (e.g. from B to C) can and should be revised.
The difference between methods “a” and “b”
The difference between methods “a” and “b” is that in the former, the measure of non-parallelism is a point estimate, for example ΔA (the difference in 4PL asymptotes). In the latter, the measure of non-parallelism is a confidence interval (CI), for example a 90% CI for ΔA.
Method a
The simplest to understand is method “a”. Here’s how it works if the measure of non-parallelism is ΔA:
- Gather some “good” historical data and calculate ΔA for each reference-test pair.
- For each ΔA, calculate the absolute value – for example, if ΔA = -0.29, the absolute value is 0.29.
- Choose what the pass rate for “good” assays and samples should be – say 99%.
- Set the upper limit for ΔA at the 99th percentile (or whatever pass rate you have chosen) of the absolute values. The lower limit for ΔA is the negative of this.
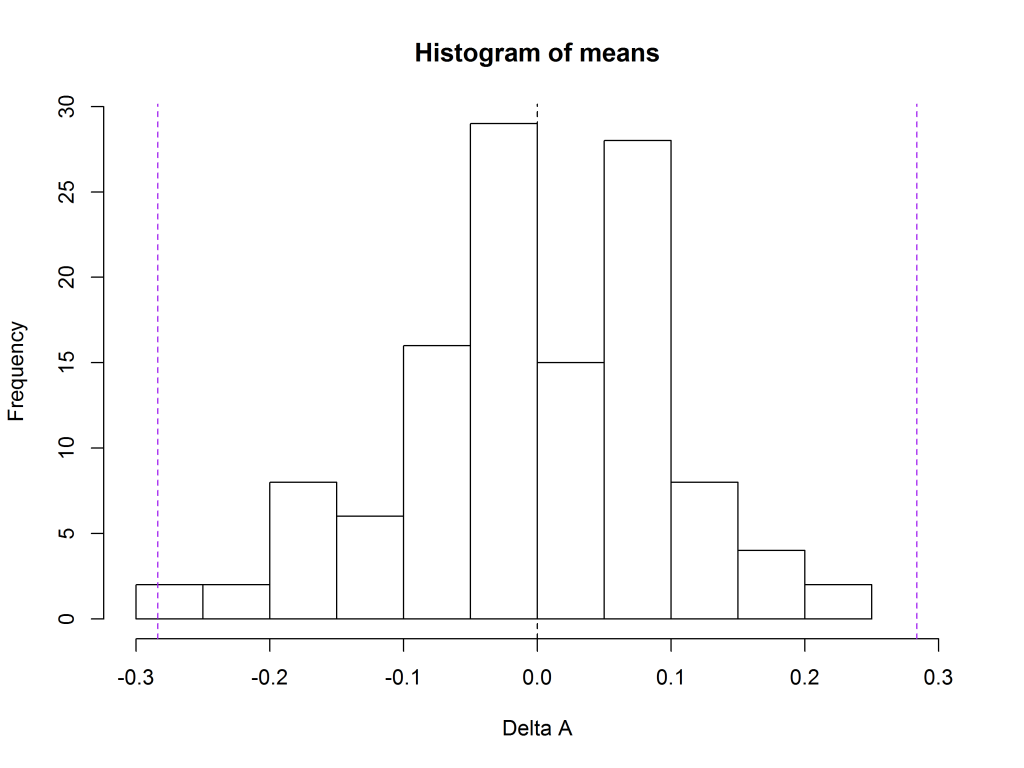
For the data in Figure 1, the 99th percentile turns out to be 0.29, so the suitability criteria on delta A are -0.29 to 0.29. These are shown by the purple vertical lines.
Method b
The process for method “b” is very similar, but everything is done for the 90% CIs for ΔA, rather than for ΔA itself:
- Gather some “good” historical data and calculate ΔA and its 90% CI for each reference-test pair.
- Choose what the pass rate for “good” assays and samples should be – say 99% again.
- For each CI, calculate the distance of its furthest point from 0 – for example, for a CI of -0.39 to 0.07, this is 0.39.
- Set the upper limit at the 99th percentile (or whatever pass rate you have chosen) of these distances. The lower limit is the negative of this.
For the data in the Figure 2, the 99th percentile turns out to be 0.49, so the limits on the CIs for ΔA are -0.49 to 0.49. These are shown by the purple vertical lines.

Compared to method “a”, this method also has the advantage of accounting for the uncertainties on the ΔAs.
Method c
Methods “a” and “b” both rely entirely on “good” data. This means that there is no guarantee that they will do what suitability tests are supposed to do – reject assays or samples which aren’t behaving normally.
Method “c” tries to remedy this by additionally incorporating data from assays or samples that should fail. This “bad” data could include, for example, degraded test samples. The suitability criteria are then set so that most of the good assays or samples will pass, while most of the bad ones will fail.
An example is shown in Figure 3, and is constructed using the same “good” data as we looked at for method “b”, but adds data from 10 “bad” reference-test pairs (shown in red). ΔA is consistently larger for the “bad” data, so it is easy to set a limit (the purple line on the right) which distinguishes between good and bad test samples.

However, the separation between the ΔAs for good and bad test samples may not always be this clear. For example, the bad test samples may still tend to have higher ΔAs than the good test samples, but it’s impossible to choose a limit for the CI which will allow all the good ones to pass while all the bad ones fail. The best that can be done in this case is a compromise: usually suitability criteria will be chosen such that some good samples fail the test.
Method d
Method “d” has a very different approach to setting the limits. Instead of setting the limits to achieve certain pass and fail rates for good and/or bad data, it instead looks at the consequences of, for example, a lack of parallelism. The limits are set to avoid these consequences becoming too severe. An analogy might be speed limits – these aren’t set by measuring the speed of lots of cars and calculating the 99th percentile, but are (or at least should be!) based on the speed at which the risk and consequences of an accident become excessive.
Anyway, back to bioassays and method “d”.
A specific example where method d could be applied is an assay where for some reason (perhaps an edge effect), one of the test replicates is sometimes a bit low or high.
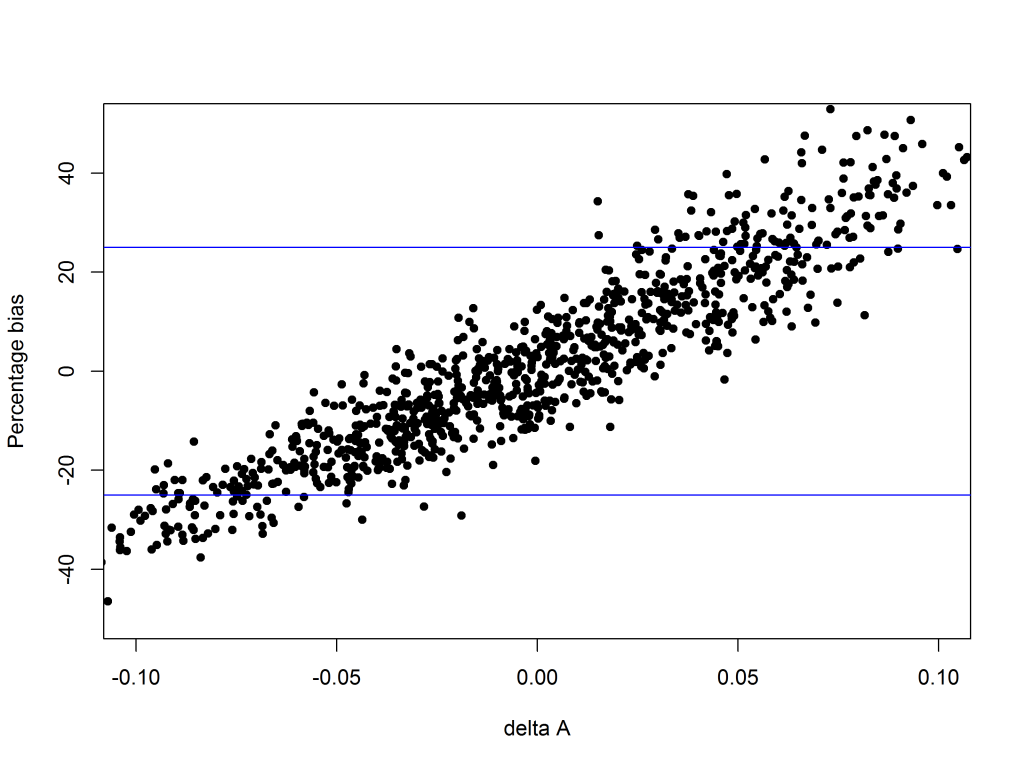
When this happens there will be non-parallelism, and in particular ΔA will be non-zero. The edge effect will also cause a bias in the relative potency. It turns out that when ΔA is negative, the bias is also negative, and vice versa, as shown in the Figure 4.
A real assay could behave similarly, though the details would be slightly different.
To set limits using method “d” in a situation like this, the first step is to decide how much bias is acceptable. This will depend on the purpose of the assay – e.g. if the assay is for batch release, the bias should be small enough that the chances of it changing the decision on whether to release a batch are small.
This in turn will depend on the release criteria and how much batch-to-batch variability there is, so deciding on the maximum acceptable bias requires a good understanding of what the assays is used for and what would happen if it was biased by a certain amount.
OK, suppose we’ve thought carefully about all this and decided that a bias of up to 25% would be OK (this limit is shown by the blue lines in Figure 4). The next step is to use this to set a limit on ΔA. This is done by using the relationship between the bias and ΔA – large values of ΔA tend to go together with large values of the bias, so if we ensure ΔA is small the bias will be small too. For this example, to restrict the bias to less than 25%, a limit of ± 0.03 on ΔA is about right. This is shown in the Figure 5 – with a limit on ΔA at ± 0.03 (the red lines), only the points shown in black will pass, and almost all of these have a bias of less than 25%.

Choosing and revising suitability methods
Each of the methods has advantages and disadvantages.
Methods “a” and “b” are the simplest, and can always be used since they only require “good” historical data, which should always be available. However, they may produce limits that don’t actually reject samples or assays that should fail – there is no way of telling since they don’t use “bad” data. Of the two, method “b” is probably better since it also accounts for the uncertainty in the suitability criterion.
Method “c” is also simple to implement, but requires “bad” historical data, from assays or samples that are known to have problems, which may not be available. Even if it is available, it may be difficult to set limits that can reliably discriminate between good and bad assays and samples. But if these obstacles can be overcome, method c is superior to methods “a” and “b” since it will ensure the suitability tests do what they are supposed to do – discriminate between good and bad assays and samples.
Method “d” is the most objective, since it is based on the real-world aim of the assay and makes the limits only as narrow as needed for the assay to achieve this aim. But it is very difficult to use in practice since it needs a detailed understanding of the assay, therefore, it is rarely used.
One final thing to mention is that suitability criteria can be revised as more data comes in, if it turns out that the original limits were either too wide or too narrow. The method used to set them can also be changed – for example if method “b” was used initially we would recommend switching to method “c” once data from degraded samples was available.






