ISS & ISE: Integrated Summary of Safety and Integrated Summary of Effectiveness
What do ISS & ISE mean?
ISS & ISE stand for integrated summary of safety and integrated summary of effectiveness, respectively. These are not merely summaries, as the name might suggest, but rather documents comprised of integrated analyses of the safety and effectiveness of a study drug [1]. In other words, the results from all clinical trials performed on the study drug are pooled together and analysed as a whole, producing combined statistical results.
When does one need ISS and ISE?
ISS and ISE are crucial aspects of New Drug Applications (NDAs), uniquely required by FDA (USA) regulation [1]. These integrated analyses are not strictly required for NDA submissions to the MHLW (Japan) and the EMA (EU), however MHLW and EMA submissions must follow the common technical document (CTD) format, which contains sections in line with ISS and ISE [2].
Key Takeaways
- ISS (Integrated Summary of Safety) and ISE (Integrated Summary of Effectiveness) are comprehensive documents that pool and analyze data from all clinical trials on a study drug. They are crucial for New Drug Applications (NDAs) in the US and are closely aligned with the sections required in the Common Technical Document (CTD) format for other regulatory bodies.
- The ISE outlines the drug’s effectiveness, including study designs, results, and dosage details. Meanwhile, the ISS focuses on overall safety, summarizing adverse events, exposure demographics, and other pharmacological assessments. These summaries must ensure all relevant data is traceable and statistically robust.
- Integrating data from studies—especially when dealing with legacy (non-CDISC) datasets—poses challenges. Early planning is essential to overcome these issues.
As of January 1st 2018, the CTD format is also mandatory for certain regulatory submissions to Health Canada including NDAs, referred to as a New Drug Submission (NDS) by Health Canada [3].
Both ISS/ISE and the sections of the CTD have essentially the same requirements, however the ISS and ISE are more rigidly formatted and require a great amount of detail of the analysis and supporting information. These integrated analyses are not required for biologic license applications (BLAs), however it is strongly recommended that they are included.
Whether for regulatory submission or not, integrated summaries are useful for:
- Discovering rare and/or unexpected trends in patient subgroups
- Improving the precision of results with a larger population size by integrating studies
- Comparing variation in study results to assess the risk and benefits
- Reaching a strong and defendable statistical conclusion
What is the common technical document (CTD)?
The CTD is an agreed, common format for the assembly of quality, safety and efficacy information for submission to regulatory authorities. This format is mandatory for NDA submissions to the EMA and the MHLW, and strongly recommended by the FDA [2]. The CTD is made up of 5 modules; module one is region specific and the remaining modules are intended to be common to all regions. The diagram below summarizes the breakdown of the CTD format, and indicates the sections equivalent to the ISS/ISE.
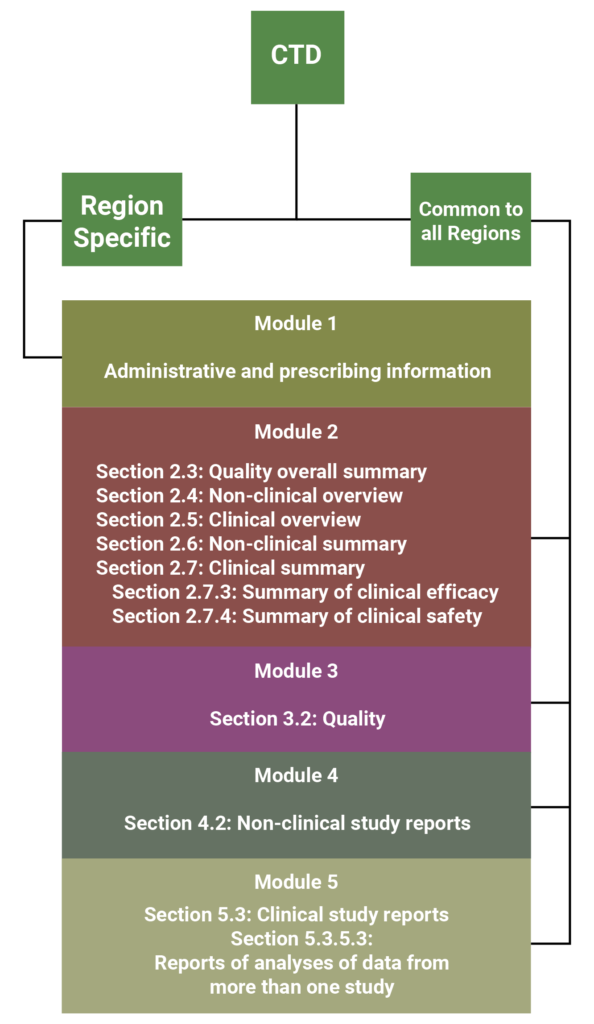
As shown by the diagram above, the FDA suggests that the best location to include the ISS and ISE in a CTD formatted application is section 5.3.5.3 of Module 5 [1]. These might also be placed in Module 2, however this rarely occurs because Module 2 is restricted to 400 pages, whereas Module 5 does not have any space limitations [1]. It might be appropriate to include the integrated summaries in Module 2 if the application is comprised of only one study or a few small studies, however the ISS and ISE often contain large appendices of tables, figures and datasets, which would not meet the 400 page requirement.
The CTD is maintained by the International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH). CTD guidelines can be found on the ICH website: http://www.ich.org/products/ctd.html.
What should an ISS and ISE include?
The ISS and ISE are critical to FDA submission, so it is important to prepare in advance and plan for the integrated summaries to aid with informed decision-making.
The aim of the Integrated Summary of Effectiveness is to define and support the nature of a drug’s effectiveness, providing all available information on the effectiveness for each claimed indication and the recommended usage.
The following are key components [4]:
- A list of individual studies with brief description of the study design and results
- Justification for any reliance on a single study
- Well demonstrated evidence of the claimed effect(s)
- Summary of evidence supporting administration
- Dosage
- Recommended dose interval
- Varied dosage for subgroups, with evidence to support the individualisation of subgroups (should be noted if specific subgroup(s) have been excluded from the trial)
- Comparison of results across multiple studies
- Identification of effects in subgroups
- Dose-response information
- Comparison with any alternative drugs
- Analysis of evidence of long-term effects, tolerance and withdrawal
- Review of results from uncontrolled studies; if relevant in supporting claims of effectiveness
The aim of the Integrated Summary of Safety is to provide an overall analysis and summary of the safety data. The following are key components [5]:
- A summary of the safety profiles from all clinical studies (can include Phase I studies in healthy volunteers; normally presented separately from study patients)
- The extent of exposure – the number of patients exposed (by gender and other demographic subgroups) and number exposed to various doses for defined durations
- Demographic and other characteristics of study population – age, gender, race, body weight, primary & secondary outcomes, and other relevant prognostic variables
- Adverse events
- Overall analysis of adverse events including event rates, deaths, adverse dropouts and other potentially serious events
- Laboratory assessments
- Summary of animal data important to human safety
- Analysis of adverse effects relating to dose-response information – This should include any evidence of dose-response variations relating to age, sex or any other subpopulations.
- Drug-drug, drug-demographic, and drug-disease interactions
- Any other pharmacologic properties – including effects on Liver, Kidneys, etc
- Long-term adverse effects and withdrawal effects
The body of the ISS and ISE are composed of text to describe study results, embedded with tables and figures of important endpoint analysis. Listings and details of statistical approaches, as well as supporting tables and figures, should be included in an appendix.
What are some potential challenges faced when producing an ISS and ISE?
In 2017 the FDA mandate for CDISC submission began; all studies from this point onwards are required to submit to CDISC (Clinical Data Interchange Standards Consortium) standards. CDISC is a global, non-profit organisation that develops data standards to help improve medical research and healthcare [6]. CDISC provides a set of standards for organising clinical trial data, including the Study Data Tabulation Model (SDTM) and the Analysis Data Model (ADaM) [6]. Since it was not mandatory to submit to CDISC standards until 2017, the majority of older studies are found in the legacy (non-CDISC) format.
This can pose a challenge for programmers, since it is likely that not every study will adhere to CDISC standards, making it more difficult to combine these datasets. Further to this, there are currently no specific CDISC guidelines for creating integrated datasets even if they all adhere to the new standards. Often grouping variables will differ, for example different studies may use different coding to group patients by age, or use different versions of the Medical Dictionary for Regulatory Activities (MedDRA) and World Health Organization (WHO) Drug Dictionary to classify drugs.
Since traceability is a key feature of a CDISC compliant submission, care must be taken to ensure that traceability is maintained throughout this process. It is important to document the process of converting datasets to SDTM and ADaM standards. It is also important to include all predecessor variables in the datasets and document them in a define.xml file, as well as ensure variables are consistent after integrating.
How can we overcome the challenges of performing an ISS and ISE?
Quantics have a good understanding of the challenges discussed above. In this section we present our recommendations for what makes a good ISS and ISE and how to prepare during the early clinical phases.
When preparing for an ISS and ISE during the early stages of a clinical trial, usually coinciding with preparation for Phase II, one should not only consider the content of the integrated summaries but also consider creating a plan to ensure optimal traceability, and the accuracy and consistency of information. One should consider the following:
- Guidelines
- Up-to-date throughout (CTD, CDISC, FDA guidelines, etc.)
- Traceability
- All data and information produced throughout the study is important to the final safety and efficacy statements, so traceability is crucial
- All information should be traceable from raw data through to summaries
- Quality Control (QC) review throughout, not just at the end
- Resource availability
- Are studies fully specified? (i.e. protocols, SAPs, annotated CRFs, coding dictionaries, define.xml, etc.)
- Prepare summary templates
- These will undergo several reviews and updates as the study progresses and product knowledge increases
- Similarity of trials for pooling
- study design, populations, etc.
- Converting study data to CDISC format
- Consider converting earlier studies for consistency and make pooling studies easier
- Avoid repetition
- e.g. information provided in summary of clinical trials is not repeated
- Statistical analysis plan
- Include all relevant trials, whether positive or negative
- Methods are detailed and easily coupled with the results
- Provide considerations; inconsistencies, further exploration, etc.
Resources
[1] FDA Center for Drug and Biologics Evaluation and Research (CDER and CBER). (April 2009). Guidance for Industry: Integrated Summaries of Effectiveness and Safety: Location Within the Common Technical Document. Retrieved from https://www.fda.gov/downloads/drugs/guidances/ucm136174.pdf.
[2] (2018). CTD: M4: The Common Technical Document. [online] Retrieved from http://www.ich.org/products/ctd.html.
[3] ca. (2018). Updated Notice – Mandatory use of the Electronic Common Technical Document (eCTD) format – Canada.ca. [online] Retrieved from https://www.canada.ca/en/health-canada/services/drugs-health-products/drug-products/activities/announcements/notice-mandatory-use-electronic-common-technical-document-ectd-format.html.
[4] FDA Center for Drug and Biologics Evaluation and Research (CDER and CBER). Integrated Summary of Effectiveness: Guidance for Industry. October 2015. Retrieved from https://www.fda.gov/downloads/drugs/guidances/ucm079803.pdf.
[5] Food and Drug Administration. (1988). Guideline for the format and content of the clinical and statistical sections of new drug applications. FDA, US Department of Health and Human Services Rockville, MA, USA. Retrieved from https://www.fda.gov/downloads/Drugs/…/Guidances/UCM071665.pdf
[6] (2018). CDISC Standards in the Clinical Research Process. [online] Retrieved from https://www.cdisc.org/standards.