
Compartmental PK Models: PK/PD Analysis II
In previous blogs, we’ve examined the statistics behind Non-Compartmental PK/PD analysis. The existence of the Non-Compartmental Analysis (NCA) implies the existence of compartmental analysis, and, indeed, it is among the most commonly used methodologies for PK/PD statistics.
Rather than extract results from data alone as in an NCA, a compartmental analysis involves fitting a model of the movement of a drug through the body to the collected data. This is done by dividing the body into theoretical “compartments” which the drug can flow into and out from. A compartmental analysis can use an unlimited number of compartments, but most PK behaviour can be well described by one or two compartments.
In this blog, we’ll focus on the simpler case of one-compartment models, before tackling two compartment and other more complex models in future editions.
What’s different about a Compartmental analysis?
Key Takeaways
- Compartmental pharmacokinetic (PK) analysis involves fitting differential equations to model how a drug moves through one or more theoretical compartments in the body, allowing for the calculation of key PK metrics such as Cmax and Tmax from the modelled concentration-time curve.
- Different administration routes (e.g. IV bolus, IV infusion, oral dose) lead to distinct model equations and concentration-time behaviours. Assumptions about absorption and elimination kinetics, typically zeroth or first order, determine the shape of the curve and influence interpretations of bioavailability and distribution volume.
- Compartmental models provide a deeper mechanistic understanding and support advanced analyses like population PK, but they require complex mathematical solutions and specialist tools (e.g. NONMEM). This makes them more suitable for later-stage drug development compared to simpler non-compartmental analysis.
Unlike a NCA, which calculates the metrics of interest directly from collected data, a compartmental analysis instead fits a model to that data to describe the full concentration-time curve. PK metrics such as maximal concentration (Cmax) and the time of maximal concentration (Tmax) can then be calculated from the fitted model.
We do this using differential equations: mathematical formulae which tell us about the rate at which a quantity changes with respect to another, in this case time. The solution to our differential equations is a function of concentration with respect to time: this is our concentration-time curve!
The process of fitting a compartmental model looks something like this:
- Decide on the number of compartments.
- For each compartment, set up a differential equation which describes the flow of the drug in and out of the compartment. This can take several forms depending on how the drug is administered – we’ll examine this in detail below. Parameters of the differential equation specific to the drug in question are typically determined from literature sources.
- Solve the differential equation to give a concentration-time curve.
- Calculate/read off metrics of interest from the calculated concentration-time curve.
While, in theory, the differential equations can be solved by hand – we examine some simple cases where this could be possible – numerical methods using software like NONMEM are typically used.
A quick note about notation here: any symbol with a dot – e.g. \(\dot{A}\) – represents the rate of change of that quantity with time.
Building a compartmental model
So, our goal in a compartmental PK analysis is to set up a differential equation to each compartment to represent the flow of our drug through the body, and then solve it to find the concentration-time curve. To do this, we need to think about how a drug moves in and out of a compartment – the kinetics of the drug flow. This can be described by the following:
\(\dot{C} = \text{Rate of Drug In} – \text{Rate of Drug Out}\)
The simplest assumption we can is that the flow is constant with time, independent of the drug concentration in the compartment. This is known as zeroth-order kinetics. As a differential equation, this would be:
\[\dot{C} = \pm k\]
Where C is the concentration of the drug and k ≥ 0 is a constant which defines the flow rate.
If \(\dot{C}\) is positive, this represents flow into the compartment, and if \(\dot{C}\) is negative, this gives flow out of the compartment. An example of zeroth-order kinetics might be an IV infusion, where a drug is administered directly into the bloodstream at a constant rate over a period of time.
The concentration-time curve for zeroth-order kinetics – the solution to the differential equation – is a straight line of gradient ±k:
\[C(t)=\pm kt + C_0\]
where C0 is the initial concentration of the drug in the compartment. In the case of flow into the compartment, we can typically assume C0 = 0.
More typically, the flow is better modelled by first-order kinetics, also known as the Law of Mass Action. Here, the flow is dependent on the concentration of drug in the compartment. Much of the movement of a drug around the body depends on concentration gradients, so it makes sense that this is among the most frequently used model of flow between compartments. The differential equation for first-order kinetics takes the form:
\[\dot{C} = \pm k t\]
As before, the sign of \(\dot{C}\) depends on whether the drug is moving into or out from the compartment. This is a slightly more complex differential equation to solve, but it can be shown that this gives exponential change in the concentration in the compartment. The concentration-time curve, therefore, takes the form:
\[C(t) = C_0 e^{\pm kt}\]
One-compartment Models
Let’s look at what the concentration-time curve for a one-compartment model looks like for a few common scenarios. We’ll assume that, for all of these, the elimination phase obeys first order kinetics, while the kinetics of the absorption phase will depend on how the drug is administered.
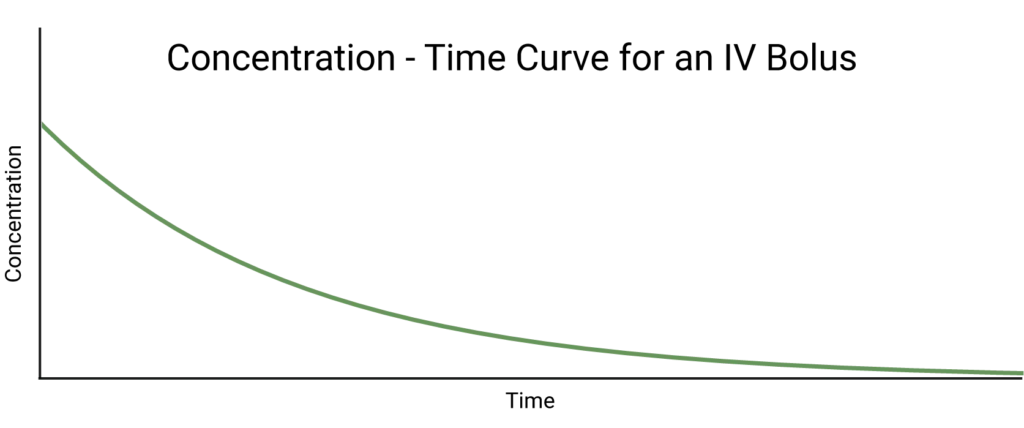
Perhaps the simplest case is a one-compartment model where a dose of a drug is administered as an IV bolus, that is, directly into the bloodstream all at once. This means that we can assume there is no absorption phase and the drug distributes instantaneously: all we observe is elimination. We assume this follows first order kinetics, so we can state that:
\[\dot{C} = -k_e C(t) \rightarrow C(t)=C_0 e^{-k_e t}\]
where \(k_e ≥ 0\) is the elimination rate constant. This tells us that the concentration of drug in the compartment decays exponentially from its maximum of C0 at t=0. We can, therefore, conclude that Cmax = C0, and Tmax = 0.
What about an IV infusion?
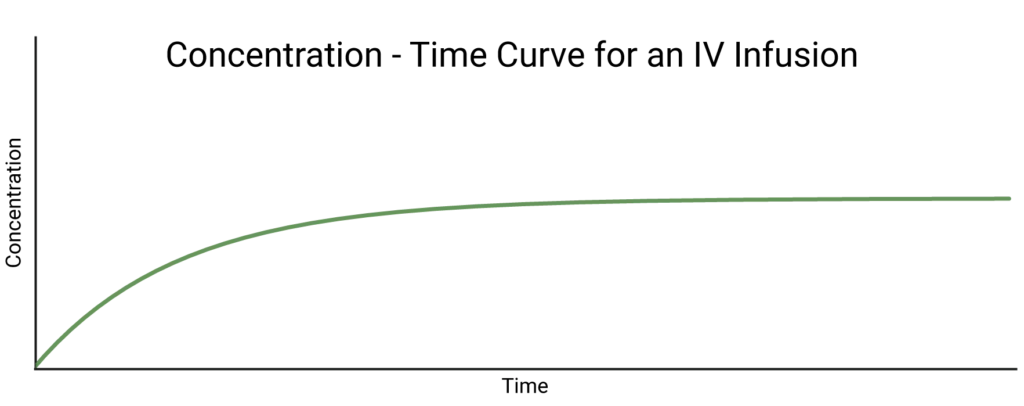
What about an IV infusion? We know, from earlier, that this means the absorption phase follows zeroth order kinetics – the drug flows into the compartment at a constant rate, which we’ll call \(k_a\). That means that, if there was no flow out of the compartment, the concentration of drug would increase linearly forever.
Recall, however, that first order elimination is ongoing at the same time as this zeroth order absorption. To get the differential equation which describes the overall behaviour of the concentration, all we need to do is add the differential equations for each individual process. This means we expect to have two terms: \(\dot{C}_a\), which describes changes in concentration due to absorption, and \(\dot{C}_e\), which describes those due to elimination:
\[\dot{C} = \dot{C}_a + \dot{C}_e = k_a – k_e C(t)\]
The mathematically inclined can prove, assuming that C = 0 at t = 0, that the solution to this differential equation is:
\[C(t)=\frac{k_a}{k_e}(1-e^{-k_e t})\]
Non-IV routes
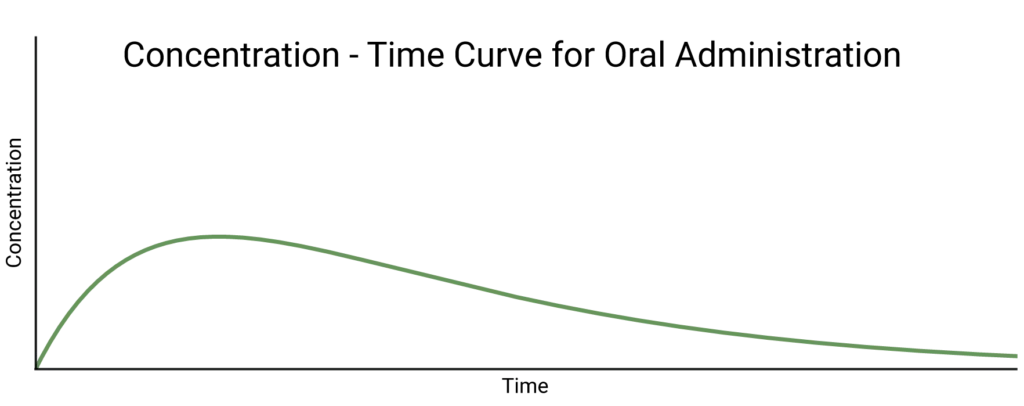
When the dose is administered by a method other than IV, we expect the absorption to follow first order kinetics. We, therefore, observe an exponential absorption phase in the concentration-time curve as well as the exponential elimination phase.
Importantly, it is likely that some of the drug will be lost in the absorption phase, for example, not all of an oral dose will make it through the gut wall into the bloodstream. How well the dose is absorbed is known as its bioavailability. An IV dose always has a bioavailability of 1.
The concentration-time curve for this case is defined by:
\[C(t)=\frac{FD}{V_d}\frac{k_a}{k_a-k_e}(e^{-k_e t}-e^{-k_a t})\]
where F is the bioavailability parameter, D is the mass of drug in the initial dose, and \(V_d\) is the volume of distribution, defined by:
\[V_d = \frac{\text{Mass of drug in body}}{\text{Concentration of drug in blood}}\]
Compartmental or NCA?
While somewhat more complex than an NCA, a compartmental PK analysis provides the benefit of a more comprehensive understanding of the PK behaviour of a drug. This can be important for later stages of the drug development process. Notably, compartmental models can be used for population PK (PopPK) analyses which account for patient covariates. This is not possible for NCA studies.
However, the added requirement for solving often complex differential equations is a drawback. Finding solutions to differential equations even numerically can be time-consuming and resource intensive, and while software solutions such as NONMEM exist, they often take significant training to use effectively. Compartmental PK analysis is, therefore, a powerful solution at later stages of drug development when a greater time and resource investment can more easily be justified.
Need help with your PK/PD Study? Find out how Quantics can help!






