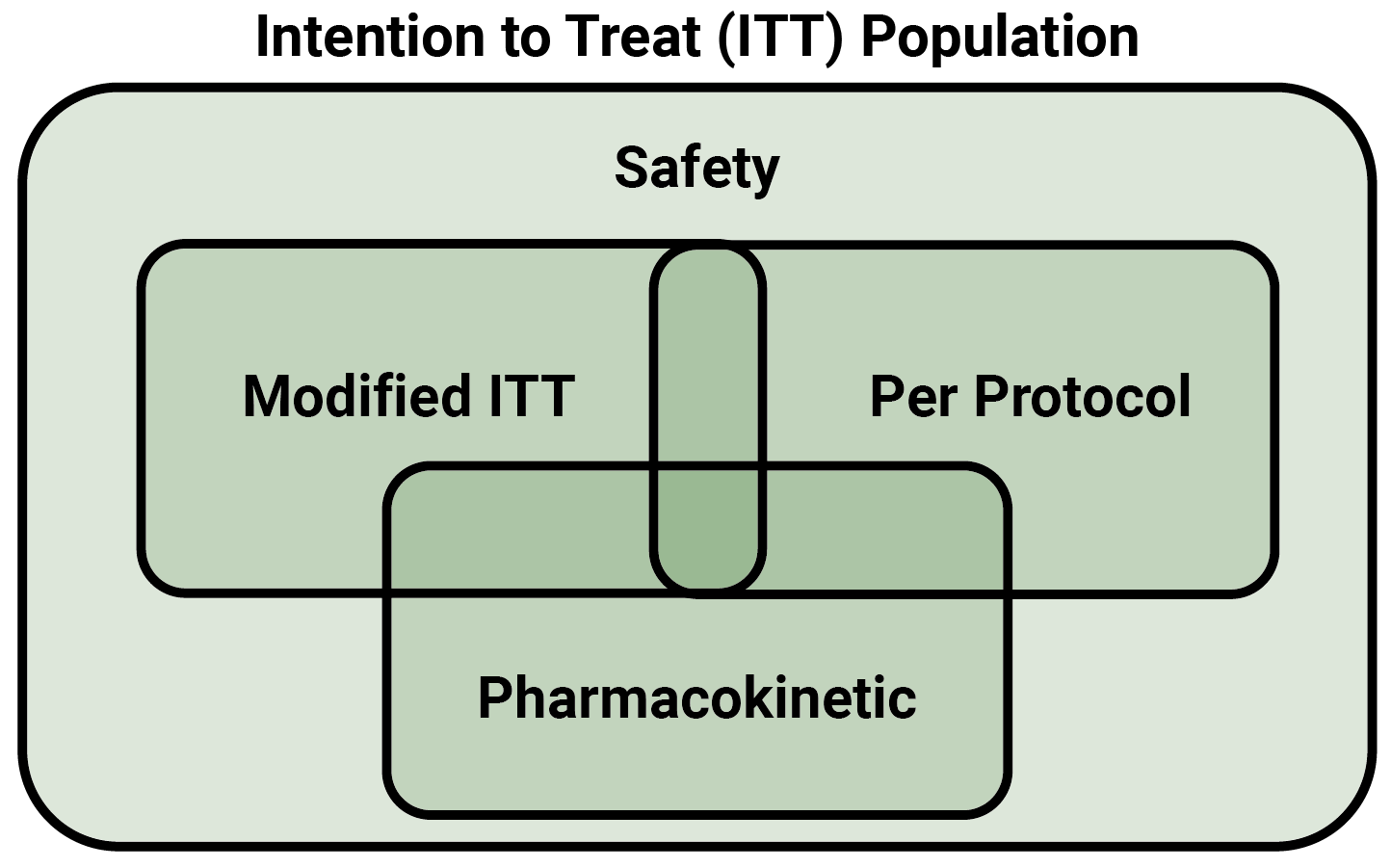
What is Bioequivalence?
One of the greatest challenges in the development of new drugs and therapeutics is that it’s just plain expensive. The average spend on a drug which passes clinical trial is on the order of billions of dollars – and don’t forget that more than 90% of drug candidates don’t make it to market – meaning much of that investment is lost. It makes sense, then, that many new drugs are expensive: innovation and research in the pharmaceutical industry needs to be funded somehow. Eventually, there comes a time where less expensive drugs are required, such as when a previously innovative treatment becomes routine. This is the realm of generics – versions of a drug which are outside of patent, and so can be manufactured by any company. But there is a problem: how do we know that a generic version of a drug is as effective and safe as the original? We could just run another clinical trial, but these are expensive and have a high failure rate, so we want to avoid one if at all possible. This is where bioequivalence comes in.
Key Takeaways
- Bioequivalence studies provide a faster, cheaper route than full clinical trials for bringing generics and new delivery routes to market, while still demonstrating that products are safe and effective.
- Two products are considered bioequivalent when they share the same active ingredient and show sufficiently similar absorption rate and overall exposure (bioavailability) at the site of action.
- Average Bioequivalence (ABE) focuses on comparing mean PK measures such as AUC and Cmax, whereas Population Bioequivalence (PBE) also factors in variability, with different regulators placing different emphasis on each approach.
The importance of bioequivalence (BE) arose in the 1980s as many of the first innovator therapeutics began to reach the end of their patent windows. This saw the rise of generics. By 2009, generic prescriptions made up 2/3 of those filled in the US, but represented just 13% of the costs. BE presents a vital methodology to ensure the safety and efficacy of these generics without requiring expensive clinical trials, reducing the cost of the therapeutic for the end user.
In addition to generics, BE studies are often used to prove that a novel delivery method or dosage of a therapeutic is safe and effective without the need for a full clinical trial. This can be potentially game changing – an oral dose of a drug previously administered, say, intravenously can make access to the treatment far easier for those without easy access to a hospital.

So, BE is vitally important for improving access to therapeutics by removing the need for expensive clinical trials. But how does it work? In short, the goal of a BE study is to show that the product under investigation behaves identically – or near enough – to a previously approved product when used as a therapeutic. The logic is that if the original therapeutic is accepted to be safe and effective, and the new therapeutic behaves identically, the new therapeutic must also be safe and effective. We can effectively “piggy-back” off the results of the original clinical trial to avoid having to repeat the trial for the new product.
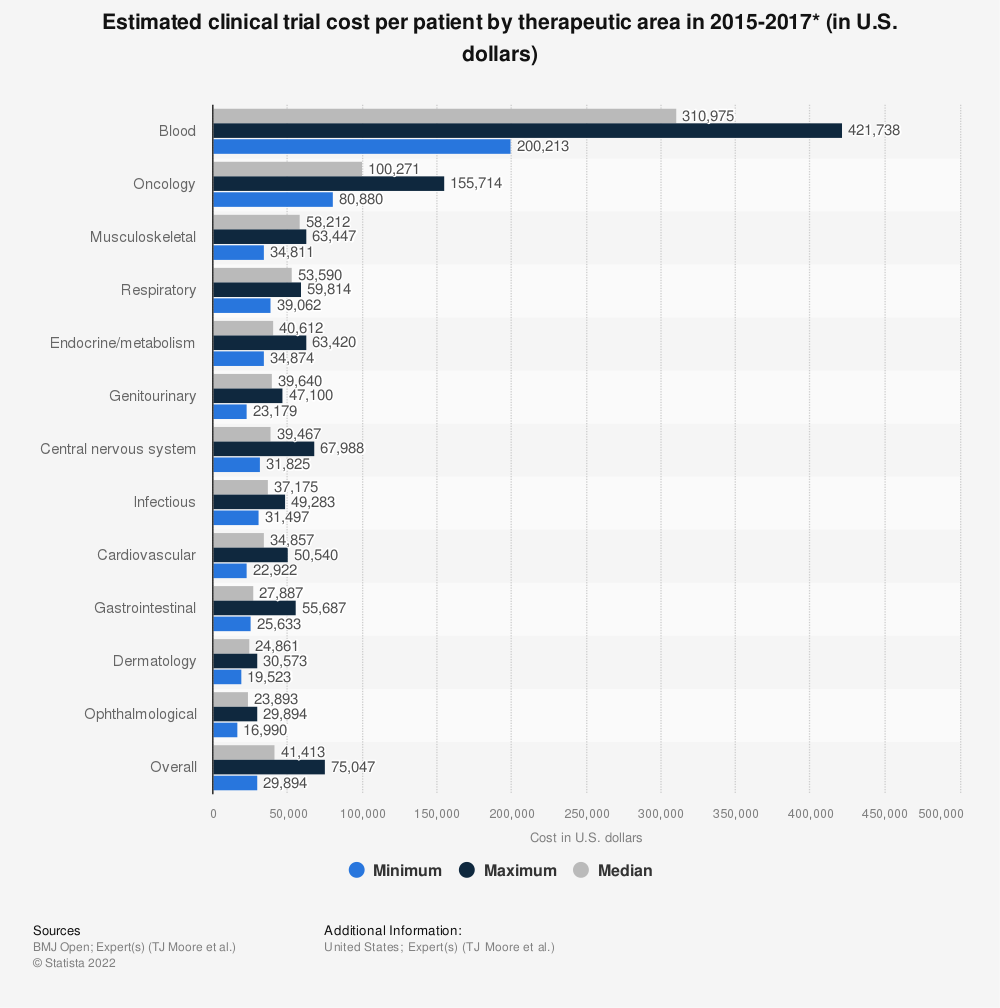
This is far faster and less expensive than a typical licensing process. For example, in the US – under the jurisdiction of the FDA – a novel therapeutic can apply for regulatory approval under a New Drug Application, or NDA. As shown in Figure 1, this can take several years or even decades from discovery to approval, including several rounds of clinical trials. By contrast, a generic drug can be approved under an Abbreviated New Drug Application, or ANDA, which cuts out the requirement for clinical trials in favour of a bioequivalence study. This not only saves time, but vast amounts of money: average per-patient costs for a clinical trial between 2015 and 2017 were found to be more than $41000, while a typical BE study tops out at $27000 per patient on average.
What does it mean, though, for two products to be bioequivalent? The FDA defines bioequivalence as “the absence of a significant difference in the rate and extent to which the active ingredient or active moiety in pharmaceutical equivalents or pharmaceutical alternatives becomes available at the site of drug action when administered at the same molar dose under similar conditions in an appropriately designed study”.
Let’s break this down. There are two key terms in this definition: pharmaceutical equivalents and pharmaceutical alternatives. The former are two drug products which contain the same active ingredient in the same dose, while the latter are products which, while they contain the same active ingredients, might contain different doses.
The condition provided by this definition is that the active ingredient must get to the target site with an equivalent “rate and extent”. These terms apply to the pharmacokinetics (PK) and bioavailability of the drug in the therapeutic, respectively.
PK describes how the drug moves through the body – we discuss this in detail here. In this case, we are particularly interested in how quickly the drug is absorbed. Bioavailability, by contrast, tells us how much of the dose of the drug actually makes it to the target site. For example, some of the dose in a nasal spray will be exhaled through the nose before it has a chance to be absorbed. By contrast, all an intravenous dose will be absorbed, so we define its bioavailability to be 1.
So, to meet the requirements for bioequivalence, two drug products must have:
- The same active ingredient
- Similar absorption rates and bioavailability of that active ingredient
But, how similar do the absorption rates and bioavailability need to be? The definition requires that there is “an absence of a significant difference”: that is, the difference is not large enough to be considered statistically significant. Ideally, the new product will be identical to the old one. But identical products won’t necessarily give identical results due to normal experimental variation. So the candidate product needs to perform similarly enough to the original that the difference is indistinguishable from statistical noise, and unlikely to render conclusions about safety and efficacy of the drug inapplicable.
Broadly speaking, there are three main ways we consider bioequivalence statistically. The first, Individual Bioequivalence (IBE), looks at an individual’s response to two products. This is often used when considering changing a patient’s individual treatment regime: a discussion for another time.
We’ll focus on the other two techniques: Average Bioequivalence (ABE) and Population Bioequivalence (PBE). Without diving into the gory details, ABE is concerned only with the average of the parameters in question with little regard given to their variability. For example, two common properties which an ABE study would examine are the Cmax and AUC (see this blog for more details about these properties). For two products to be bioequivalent according to an ABE study, the average (typically geometric mean) Cmax and AUC of the trial product across the study population would have to fall near enough to that of the original such that the difference is not considered statistically significant. For the FDA, “near enough” means within 80%-125% of the original product – a metric which “is based on a clinical judgment that a test product with [bioavailability] measures outside this range should be denied market access”.

ABE has one flaw: it is possible for, say, the (geometric) mean of a candidate product’s Cmax to fall safely within the required range while exhibiting higher variability than the original product (see this blog for more about the differences between accuracy and precision). While it is unlikely that a product approved under ABE will prove dangerous or ineffective as a result of a difference in variability, it is nevertheless worth considering.
Population Bioequivalence (PBE) does just that: it takes into account both the average and variance of BE metrics across the study population. This means that a PBE study provides evidence that a test product not only behaves similarly to an original on average, but that it exhibits a similar degree of consistency in its behaviour too.
These different methodologies are preferred by different regulators. In their guidance, the EMA refer only to ABE with little consideration for PBE. Conversely, the FDA places a far stronger emphasis of PBE in their draft updated guidance. As with most (if not all) statistical processes, the exact analysis method required will vary with the product in question. In many cases, checking the average is sufficient to ensure bioequivalence. If variability is of particular importance for a certain product, then PBE might be more appropriate.
Testing for bioequivalence, then, is a crucial process for a pharma industry where getting a drug to market is – often necessarily – difficult. It gives patients access to generic drugs without requiring they undergo full clinical trials, dramatically reducing their cost. It also allows for greater innovation in drug delivery methods – much of our BE work at Quantics involves analyses for nasal sprays. Expect to see BE become ever more prevalent as innovation progresses in the world of pharmaceuticals.
Quantics have years of experience supporting bioequivalence studies. To find out more, check out our bioequivalence services page.